当今,缺血性疾病仍具有较高的发病率和死亡率,通过治疗性血管新生可以改善这些缺血区域的血流灌注,进而挽救濒死组织。然而,已开展的许多临床试验表明促血管新生策略在缺血性疾病尤其是在缺血性心脏病的治疗中有待进一步优化。近日,复旦大学附属中山医院 心脏病全国重点实验室葛均波院士、李华研究员团队发现转录因子GTF2H4能够促进内皮细胞在缺氧微环境中发生部分内皮间质化(图1),改善缺血后微循环灌注,并详尽阐明了其潜在的分子机制,为缺血性疾病的治疗改善提供了全新的思路。
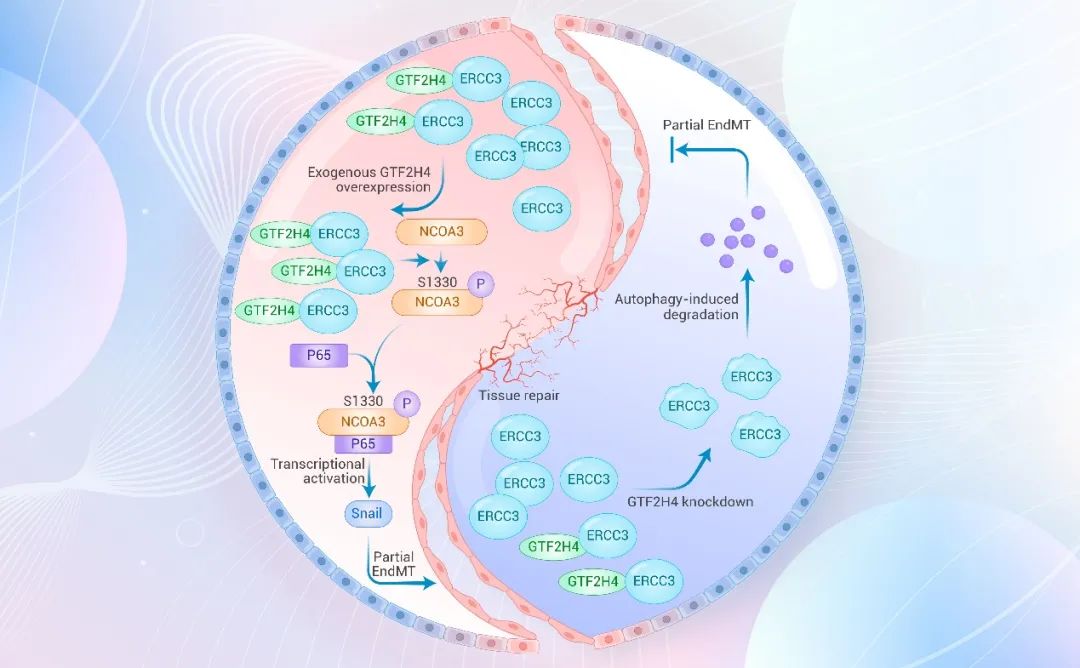
 图2
图2
缺血性疾病包括缺血性心脏病、缺血性脑疾病和外周动脉疾病,以组织器官缺血缺氧所致的血流限制或阻塞为特征。定位于血管内膜的内皮细胞最早感应缺氧信号,在组织缺血后微循环功能障碍和病理性血管新生中发挥着重要作用。内皮间质化(Endothelial to mesenchymal transition, EndMT)是指内皮细胞在TGF-β、氧化应激等刺激因素下失去内皮表征并向间充质细胞转化的过程。EndMT在心血管系统的发育中起到重要作用,也参与瓣膜病、肺动脉高血压、动脉粥样硬化等心血管疾病的发生发展。区别于完全间充质转化,部分内皮间质化是内皮细胞向间充质细胞发生转分化的中间表型,其特征表现为内皮特征的保留或部分丧失,而获得间充质表征。已有研究证实部分EndMT与血管新生具有重叠的生物学行为,间充质激活的内皮细胞具备损伤后组织修复的重要功能。
一
缺血缺氧环境诱导内皮细胞发生部分EndMT
葛均波、李华团队研究发现:在体实验中,小鼠心肌梗死后梗死边缘区内皮细胞逐渐获得α-SMA间充质标记物;体外实验中,微血管内皮细胞经缺氧处理后也获得α-SMA、Fibronectin等间充质标记物,并伴随有CD31和VE-Cadherin内皮标记物的部分丢失,内皮细胞的成管功能仍得以完全保留,提示缺血缺氧诱导内皮细胞发生部分EndMT(图3)。
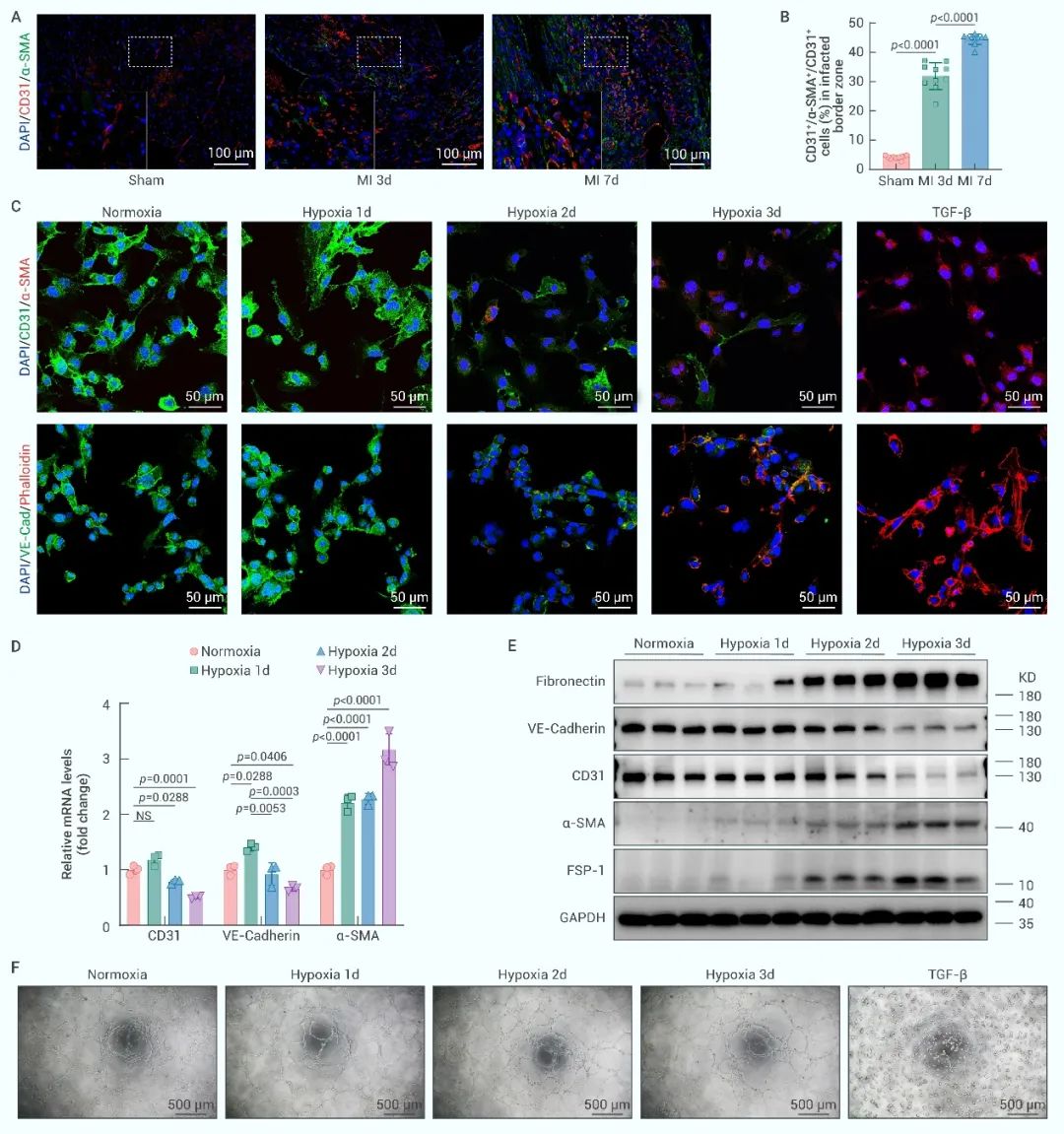
图3
二
GTF2H4缓解缺氧诱导的微血管内皮细胞损伤,并促进缺氧诱导的部分EndMT
研究团队基于前期获取的心肌梗死患者支架再通前后血液样本转录组测序结合GTEx数据库的相关研究结果,发现转录因子GTF2H4对缺氧信号显著响应,表明其在机体应对缺血缺氧损伤的反应中有着重要作用。GTF2H4作为通用转录复合体TFIIH家族的重要成员,在细胞生长、发育、代谢等方面发挥重要的调控作用。研究者随后确证了时序性缺氧与内皮细胞中GTF2H4表达水平的关联,并通过基因编辑技术证实GTF2H4能够缓解缺氧诱导的微血管内皮细胞损伤,并正向调控缺氧诱导的部分EndMT(图4)。体外缺氧环境下的功能学检测也发现GTF2H4诱导的部分EndMT增强了微血管内皮细胞的迁移能力,并促进离体动脉的血管出芽(图5)。
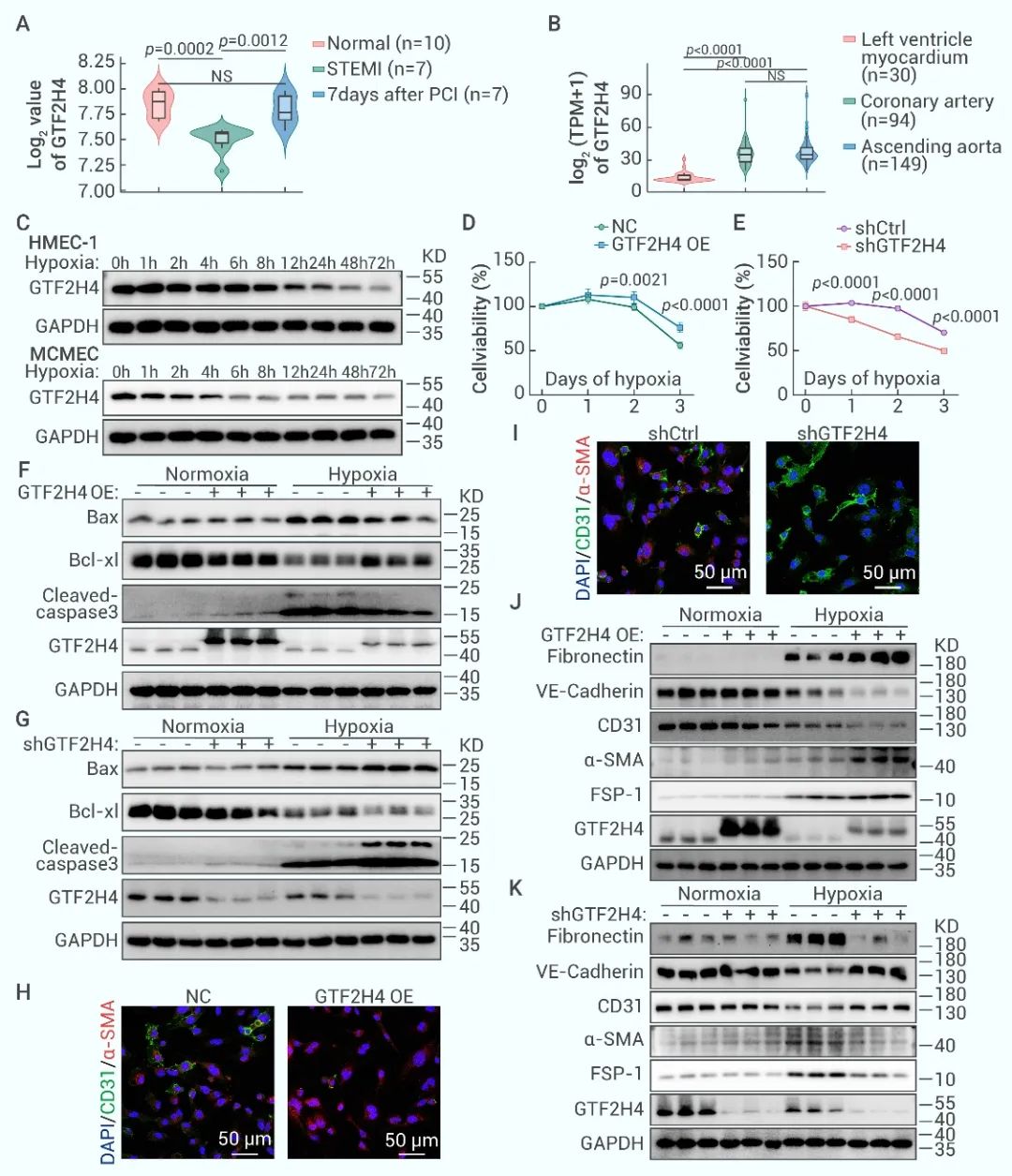
图4
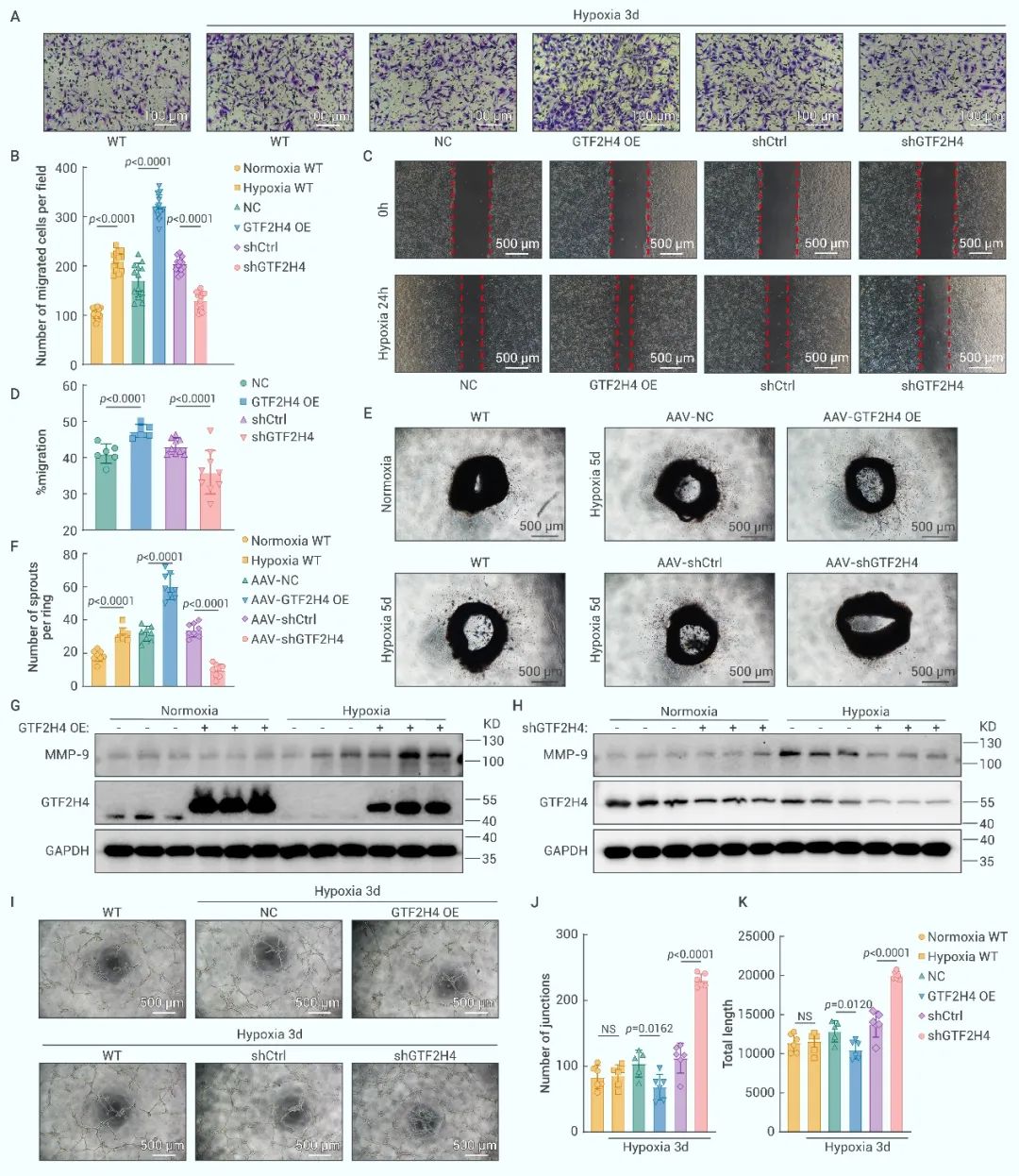
图5
三
GTF2H4通过自噬介导的降解路径调控ERCC3,二者协同调控部分EndMT
为解析GTF2H4调控部分内皮间质化的分子机制,研究团队开展4D-Label-Free蛋白质组学技术筛选得到了GTF2H4的靶分子ERCC3,此为TFIIH转录复合体上与GTF2H4毗邻的分子,具有依赖DNA的ATP酶活性和从3’端-5’端的解旋酶活性,在转录起始和核苷酸切除修复中承担着重要的作用。基于上述组学的数据结果,研究团队进行分子生物学实验验证了GTF2H4与ERCC3二者之间的互作关系,并得出GTF2H4通过自噬介导的降解路径调控ERCC3(图6)。
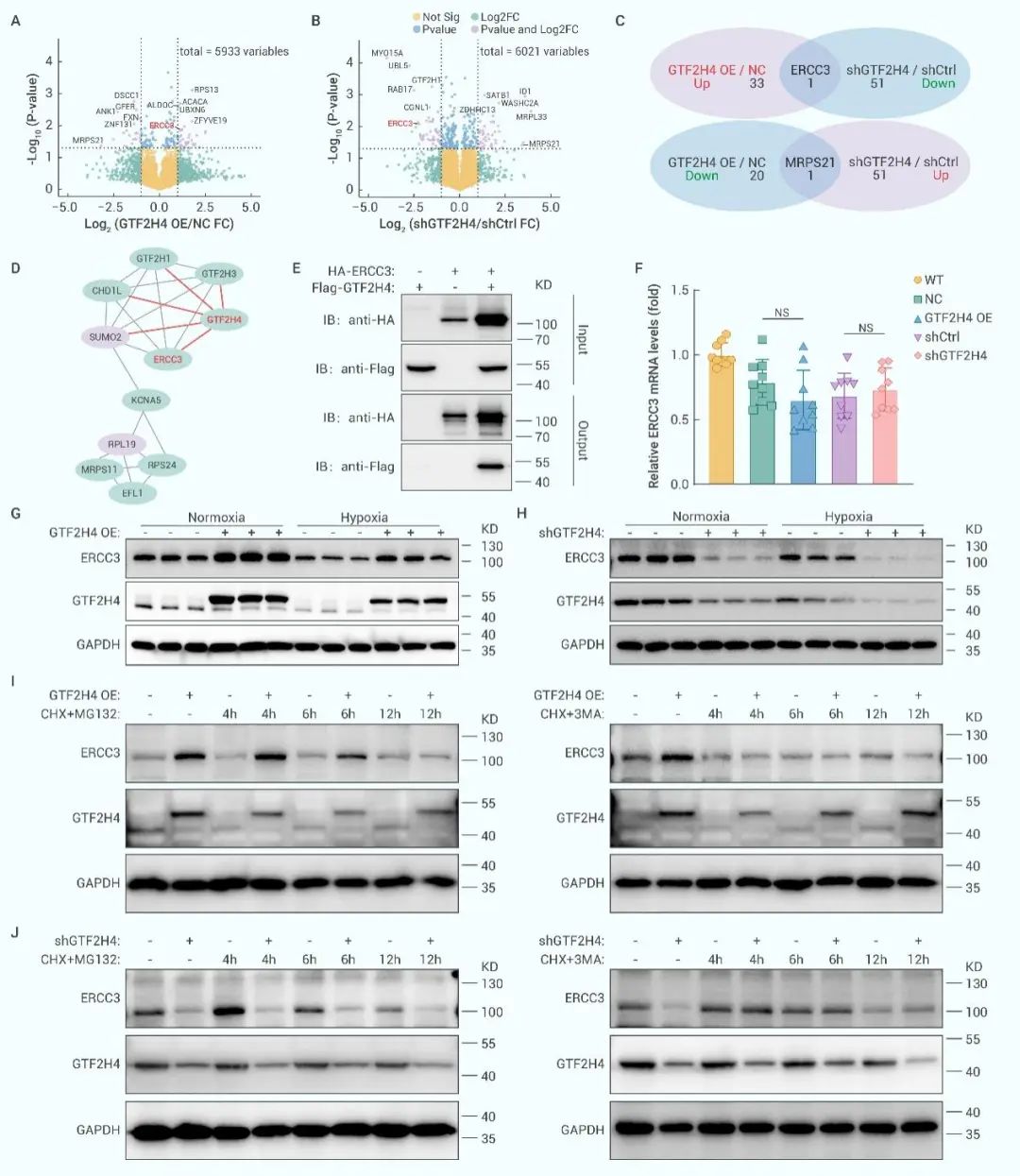
图6
随后,研究团队探讨了靶分子ERCC3对GTF2H4的反向调控及其介导的下游分子生物学功能,发现敲低ERCC3显著抑制GTF2H4的表达,并发挥与敲低GTF2H4相似的抑制部分EndMT效应,而敲低ERCC3同样也能够削弱高表达GTF2H4背景下的促间质化水平,这提示了GTF2H4功能的发挥与ERCC3存在协同效应。进一步,构建丧失ERCC3结合能力的GTF2H4突变体E310K/R314E,实验发现当GTF2H4无法结合ERCC3时,其促进部分EndMT的效应也无法发挥。由此,证实了GTF2H4调控缺氧诱导的部分EndMT依赖于其与ERCC3的结合,二者以复合体的形式协同发挥作用(图7)。
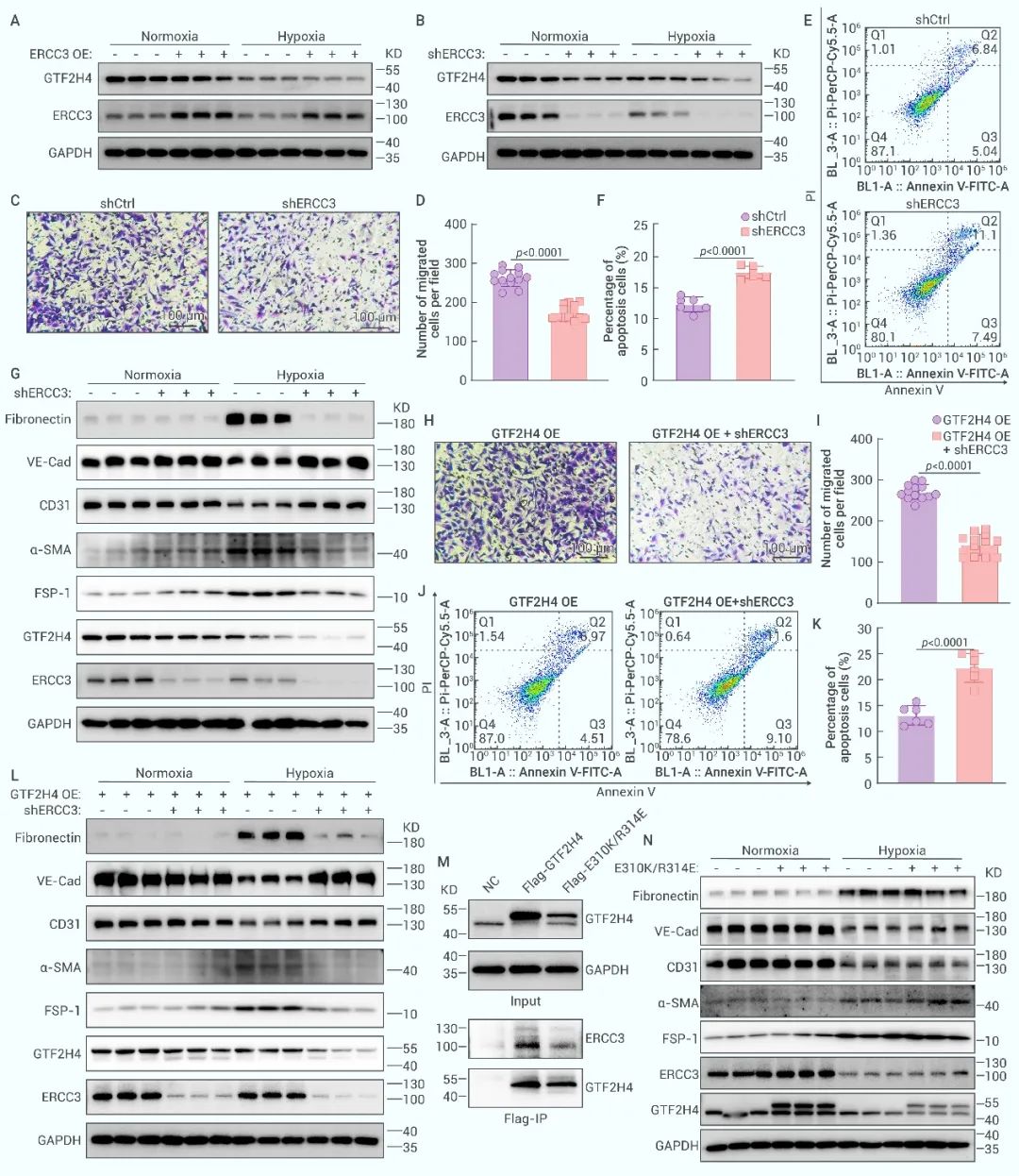
图7
四
GTF2H4通过NCOA3 S1330位点的磷酸化调控NF-κB激活的部分EndMT
为阐明GTF2H4/ERCC3调控部分EndMT的分子网络,研究团队利用4D-Label-Free磷酸化蛋白质组学联合磷酸化蛋白质通路芯片开展高通量通路富集,并通过双荧光素酶、EMA等试验得出GTF2H4主要通过NF-κB/Snail这一信号通路调控部分EndMT(图8)。
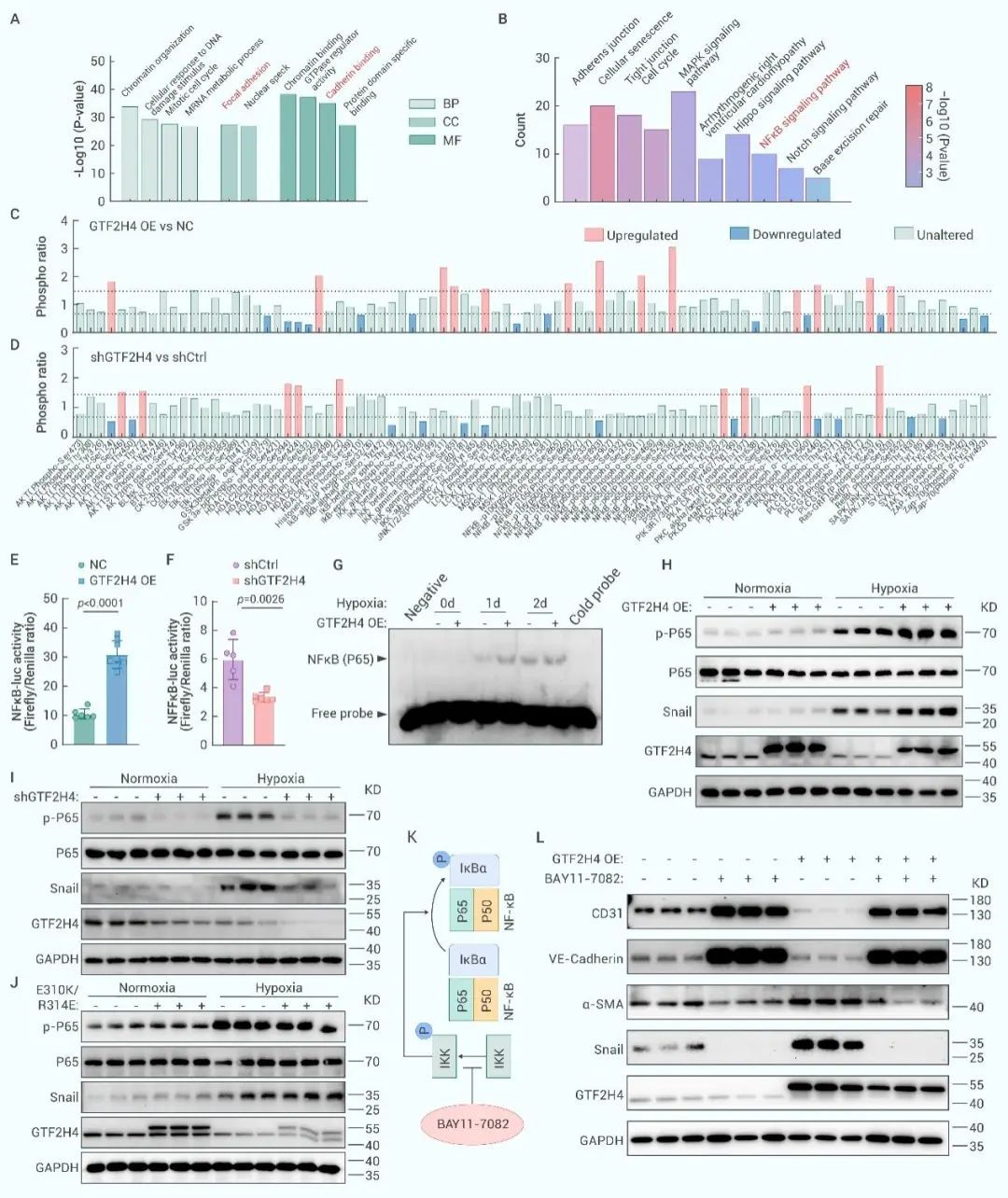
图8
深入研究中,团队发现 NCOA3 S1330位点的磷酸化位于GTF2H4过表达和敲低差异表达磷酸化肽段的交集之中。继而,针对该NCOA3的1330位点进行点突变设计,构建磷酸化失活突变体S1330A和仿磷酸化突变体S1330E/D,得出GTF2H4能够促进NCOA3上1330位点的丝氨酸发生磷酸化,进而结合p65以激活NF-κB-Snail通路,最终发生部分EndMT(图9)。
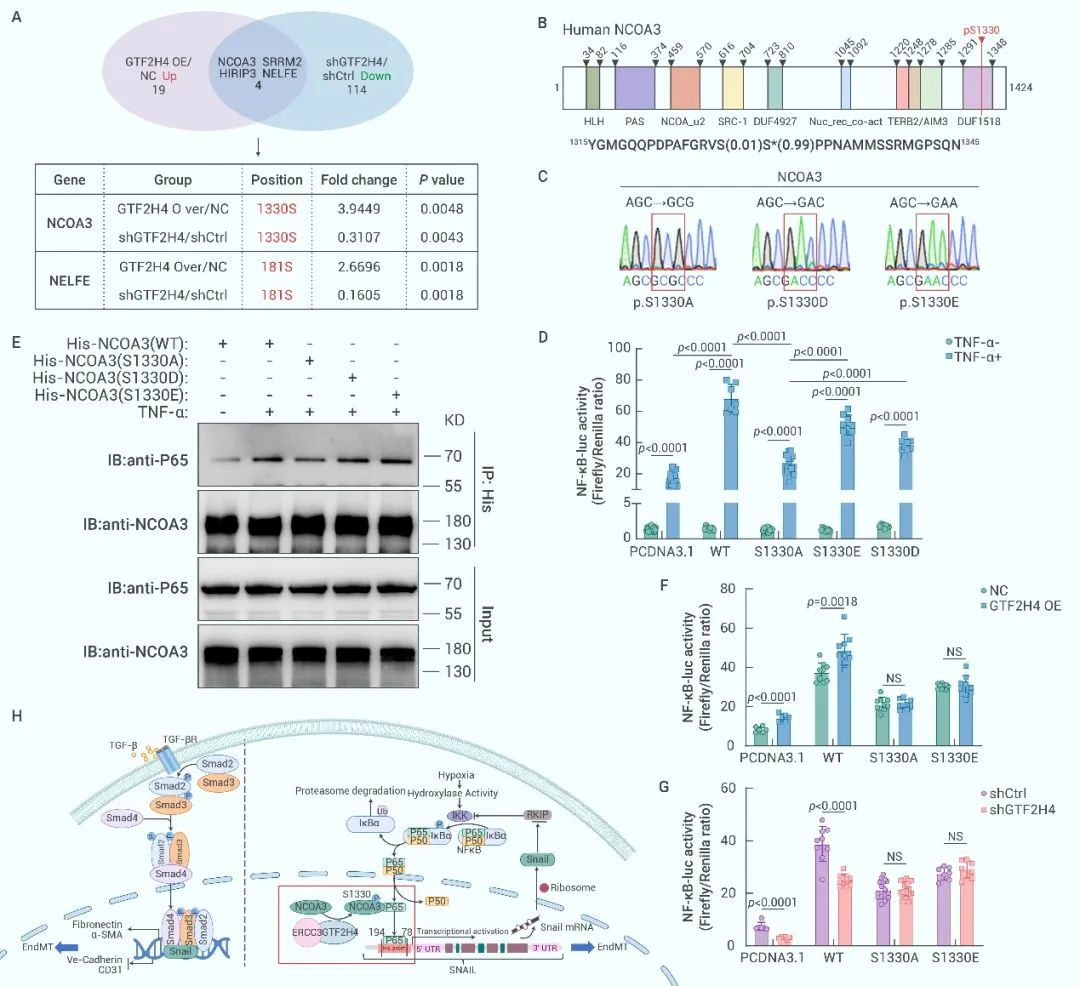
图9
五
GTF2H4促进小鼠下肢缺血后的部分EndMT以及血流恢复
动物在体研究中,研究团队利用内皮细胞靶向的AAV感染小鼠腓肠肌实现小鼠下肢内皮细胞GTF2H4高表达或敲低的基因编辑干预后,再成功构建小鼠下肢缺血模型并动态监测其下肢血流及恢复情况,发现GTF2H4能够显著改善下肢缺血后血运重建,伴随部分EndMT发生(图10)。
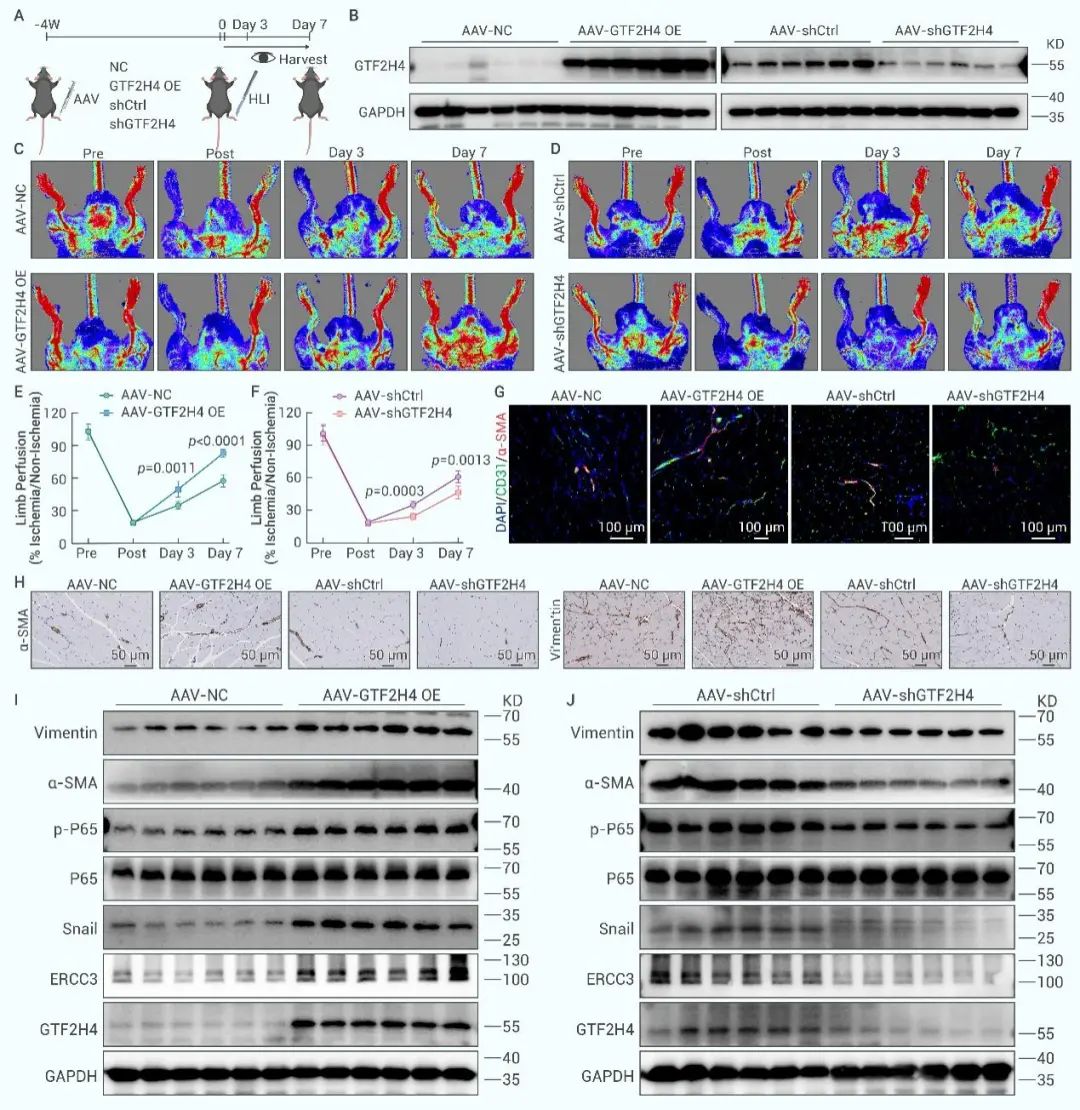
图10
该研究揭示了部分内皮间质化在缺血性疾病血管新生中的关键作用,并首次在体内外阐明转录因子GTF2H4能够通过NCOA3/NF-κb/Snail通路促进缺血缺氧诱导的部分内皮间质化,改善缺血后血运重建和组织修复。上述数据提示GTF2H4可能在缺血性疾病的靶向干预治疗中具有一定医学转化意义。
复旦大学附属中山医院心内科博士研究生方哲彦(左)为本文第一作者,葛均波院士(中)和李华研究员(右)为本文通讯作者(图11)。

图11